
Apoptosis
Creative BioMart Apoptosis Product List
Immunology Background
Background
Cell death is an essential process in the life cycle of multicellular organisms. Apoptosis, derived from the Greek word meaning "falling off," refers to a highly regulated process of cell death that allows organisms to control cell numbers and eliminate cells that threaten the integrity of the organism. Unlike necrosis, a form of traumatic cell death resulting from acute cellular injury, apoptosis is a controlled and energy-dependent process that plays a vital role in development, immune function and disease prevention.
 Fig. 1: Illustration of an apoptotic cell (Created with BioRender.com).
Fig. 1: Illustration of an apoptotic cell (Created with BioRender.com).Physiological Roles of Apoptosis
Apoptosis plays critical roles in maintaining the health and functionality of multicellular organisms. It is a tightly regulated process that is essential for various physiological functions at different stages of life. Apoptosis is essential for the proper development, functioning and maintenance of multicellular organisms. Its role in organ development, regulation of the immune system, maintenance of tissue homeostasis, and prevention of disease underscores its importance in biology.
Embryonic Development: Apoptosis is critical during embryogenesis for the formation of tissues and organs. It helps to remove excess or misplaced cells, contributing to the proper formation of structures such as fingers and toes by eliminating cells in the interdigital spaces (e.g., forming separate digits by removing ligaments). In addition, in the development of the central nervous system, apoptosis removes excess neurons to refine neural circuits, ensuring that only the necessary and properly connected neurons survive.
Immune System Regulation: During the development of the immune system, apoptosis eliminates self-reactive T and B cells in the thymus and bone marrow, respectively. This process, known as negative selection, prevents the development of autoimmune diseases by ensuring that immune cells do not attack the body's own tissues. After an immune response, apoptosis is responsible for removing activated immune cells (e.g., T cells and neutrophils) when their job is done. This prevents prolonged inflammation and potential tissue damage, contributing to immune homeostasis.
Tissue Homeostasis: Apoptosis maintains tissue homeostasis by regulating cell numbers in tissues with high cell turnover rates, such as skin, intestinal epithelium, and blood cells. It ensures that old, damaged, or unnecessary cells are removed and replaced by new cells to maintain the overall health of the tissue. Cells that acquire irreparable DNA damage, whether due to environmental factors (e.g., UV radiation) or internal stress, are eliminated by apoptosis to prevent the spread of mutations that could lead to cancer or other diseases.
Development of the Nervous System: Apoptosis plays a role in synaptic pruning during brain development. It removes excess synaptic connections, allowing the nervous system to refine and strengthen the most efficient neural pathways, which is essential for cognitive function and learning.
Hormone-Dependent Tissue Involution: After lactation, apoptosis is involved in the involution of the mammary gland, in which unneeded mammary epithelial cells are systematically removed. In the female reproductive system, apoptosis regulates the cyclic shedding of the endometrium during menstruation.
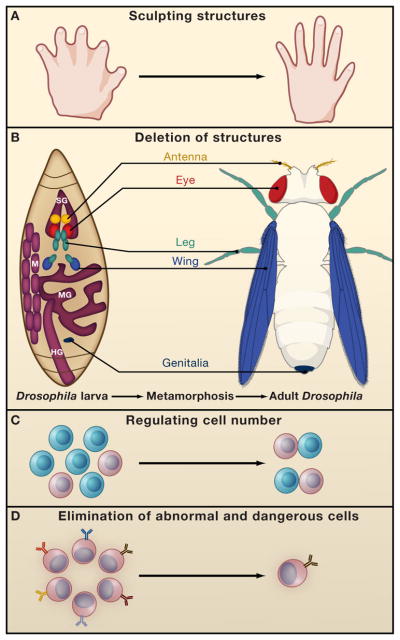 Fig. 2: Functions of PCD during development. (A) PCD regulates proper structure sculpting by eliminating interdigital webbings. (B) During Drosophila metamorphosis nearly all larval structures are destroyed such as the Salivary glands (SG), Muscles (M), midgut (MG) and hindgut (HG). (depicted by purple), whereas novel structures are raised from undifferentiated cells termed imaginal discs (depicted by various colors). The locations and developmental fates of the imaginal discs are similarly illustrated. PCD also controls cell number for example by deleting cells which fail to partner (C) and eliminates dangerous and abnormal cells such as autoreactive lymphocytes (D) (Fuchs and Steller, 2011).
Fig. 2: Functions of PCD during development. (A) PCD regulates proper structure sculpting by eliminating interdigital webbings. (B) During Drosophila metamorphosis nearly all larval structures are destroyed such as the Salivary glands (SG), Muscles (M), midgut (MG) and hindgut (HG). (depicted by purple), whereas novel structures are raised from undifferentiated cells termed imaginal discs (depicted by various colors). The locations and developmental fates of the imaginal discs are similarly illustrated. PCD also controls cell number for example by deleting cells which fail to partner (C) and eliminates dangerous and abnormal cells such as autoreactive lymphocytes (D) (Fuchs and Steller, 2011).Inducers of Apoptosis
Apoptosis can be triggered by a variety of internal and external factors, known as inducers. These inducers initiate the apoptotic process through different signaling pathways, leading to the activation of caspases and the eventual death of the cell.
DNA Damage: DNA damage can result from exposure to ionizing radiation, ultraviolet (UV) light, chemical agents (such as alkylating agents), or reactive oxygen species (ROS). DNA damage activates the tumor suppressor protein p53, which induces the expression of pro-apoptotic genes like Bax, Puma, and Noxa. These proteins promote mitochondrial outer membrane permeabilization (MOMP), leading to the release of cytochrome c and activation of the intrinsic (mitochondrial) apoptotic pathway.
Oxidative Stress: Oxidative stress results from an imbalance between the production of ROS and the ability of the cell to detoxify these reactive intermediates. Excessive ROS can damage cellular components, including lipids, proteins, and DNA. ROS can also directly trigger the release of pro-apoptotic factors from mitochondria, leading to caspases activation and apoptosis.
Growth Factor Deprivation: Cells depend on external signals, such as growth factors, to survive. Deprivation of these signals can induce apoptosis. Growth factor deprivation leads to the inactivation of survival pathways, such as the PI3K/Akt pathway, which normally suppresses apoptotic signaling. Without these signals, pro-apoptotic factors are activated, leading to cell death.
Cytotoxic Drugs: Many chemotherapeutic agents induce apoptosis as part of their anticancer effect. These drugs include DNA damaging agents (e.g., doxorubicin, cisplatin), antimetabolites (e.g., 5-fluorouracil), and microtubule inhibitors (e.g., paclitaxel). Cytotoxic drugs can induce apoptosis by a variety of mechanisms, including direct DNA damage, disruption of mitotic spindle function, and inhibition of key survival pathways. These effects converge on the activation of intrinsic or extrinsic apoptotic pathways.
Hypoxia: Hypoxia, or low oxygen levels, is a common stress condition that can occur in various pathological situations, including ischemia and tumor growth. Severe hypoxia can lead to the stabilization of hypoxia-inducible factors (HIFs), which can upregulate pro-apoptotic genes. In addition, hypoxia can lead to mitochondrial dysfunction and the generation of ROS, triggering apoptosis.
Immune Cell-Mediated Induction: Cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells induce apoptosis in infected or cancerous cells as part of the immune response. CTLs and NK cells release perforin, which creates pores in the target cell's membrane, allowing the entry of granzymes. Granzymes, particularly granzyme B, directly activate caspases within the target cell, initiating apoptosis.
Oncogene Activation: Oncogenes like MYC, when overexpressed or constitutively activated, can induce apoptosis as a fail-safe mechanism to prevent uncontrolled cell proliferation. Oncogene-induced apoptosis often occurs through the activation of the p53 pathway or through the upregulation of pro-apoptotic Bcl-2 family members, leading to mitochondrial-mediated apoptosis.
Viral Infections: Certain viral infections can induce apoptosis to limit viral replication or as a result of viral manipulation of the host cell machinery. Some viruses activate apoptotic pathways to evade immune detection, while others inhibit apoptosis to prolong host cell life. For example, the HIV virus can induce apoptosis in CD4+ T cells, contributing to the exhaustion of the immune system.
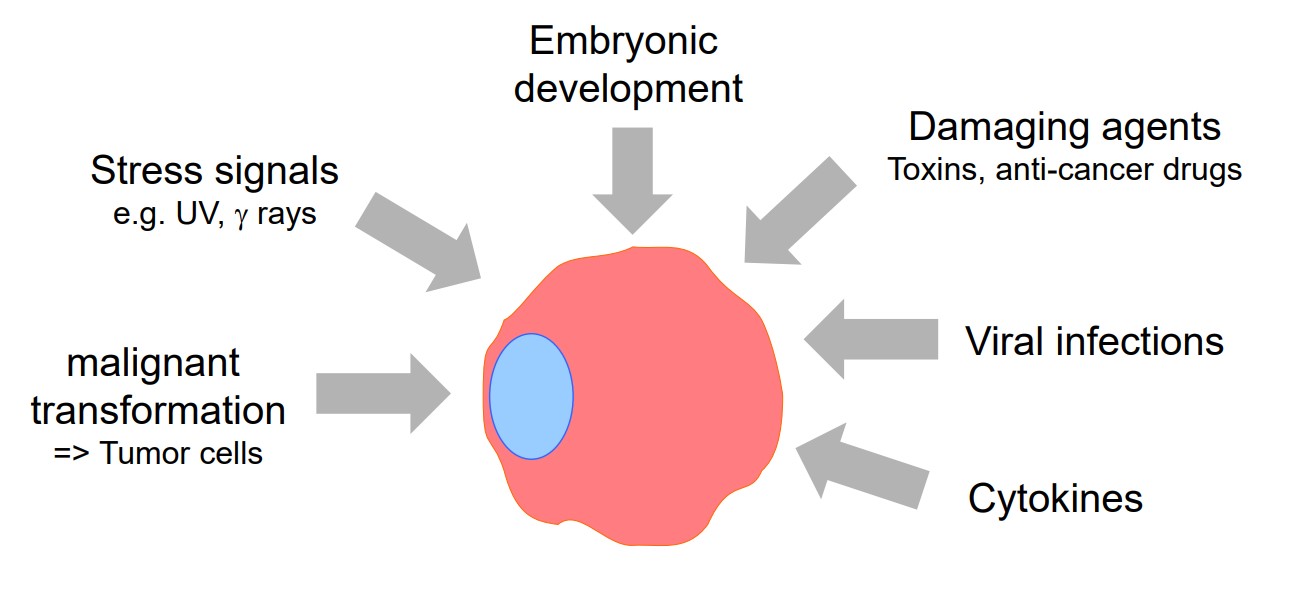 Fig. 3: Factors induce apoptosis.
Fig. 3: Factors induce apoptosis.Stages of Apoptosis
Apoptosis involves a series of well-orchestrated events that lead to the systematic dismantling of cellular components. The process can be divided into several key stages:
Initiation: Apoptosis can be triggered by intrinsic or extrinsic signals. Intrinsic signals often arise from within the cell, such as DNA damage, oxidative stress, or mitochondrial dysfunction. Extrinsic signals are typically external, such as the binding of death ligands to cell surface receptors.
Execution: The execution phase is characterized by the activation of caspases, a family of proteases that serve as the main executors of apoptosis. Once activated, caspases cleave specific substrates within the cell, leading to the breakdown of the nuclear envelope, cytoskeleton, and cellular organelles.
Degradation: After caspase activation, the cell undergoes morphological changes, including cell shrinkage, chromatin condensation, and membrane blebbing. Finally, the cell fragments into apoptotic bodies, which are small membrane-bound vesicles containing cellular components.
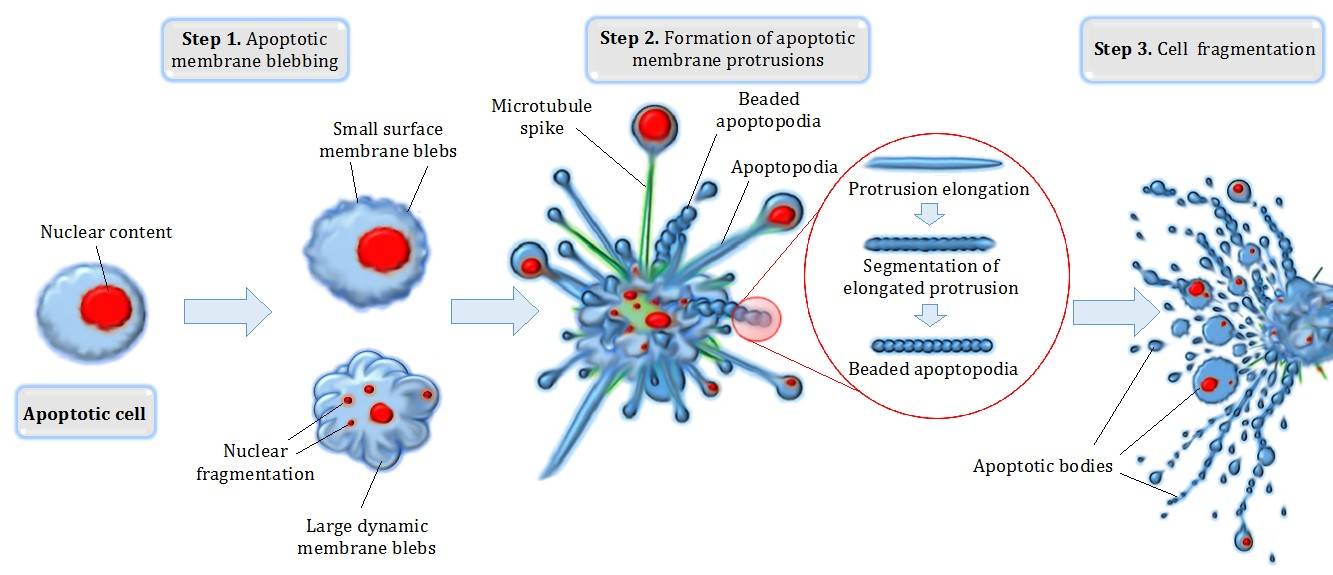 Fig. 4: Different steps in apoptotic cell degradation.
Fig. 4: Different steps in apoptotic cell degradation.Phagocytosis: Apoptotic bodies are rapidly recognized and engulfed by neighboring phagocytic cells, such as macrophages. This ensures that the cellular debris is efficiently removed without triggering an inflammatory response.
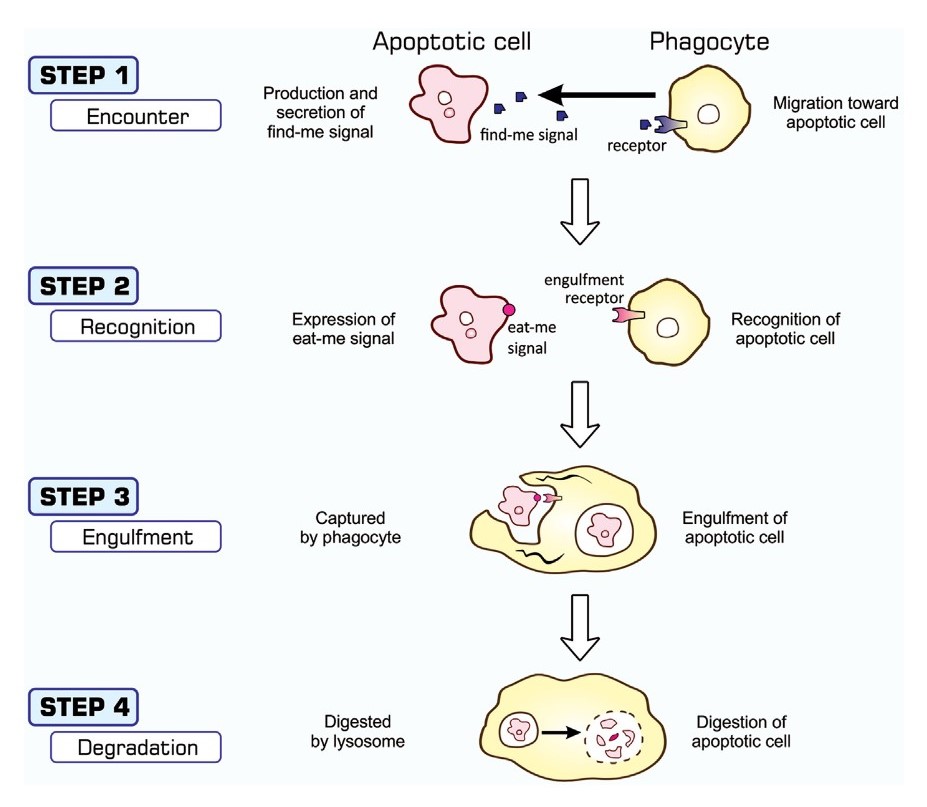 Fig. 5: Processes of apoptosis-dependent phagocytosis. Cells undergoing apoptosis secrete substances that attract phagocytes (STEP 1), often called find-me signals, and simultaneously express eat-me signals on their surface (STEP 2). Phagocytes that come into close proximity to apoptotic cells recognize and bind eat-me signals using engulfment receptors (STEP 2) and activate signaling pathways for the induction of phagocytosis. The culmination of this signal transduction is the generation of pseudopodia that help phagocytes surround and incorporate apoptotic cells (STEP 3). Materials engulfed exist as phagosomes, which subsequently fuse with the lysosomes for degradation (STEP 4) (Nainu, Shiratsuchi, and Nakanishi, 2017).
Fig. 5: Processes of apoptosis-dependent phagocytosis. Cells undergoing apoptosis secrete substances that attract phagocytes (STEP 1), often called find-me signals, and simultaneously express eat-me signals on their surface (STEP 2). Phagocytes that come into close proximity to apoptotic cells recognize and bind eat-me signals using engulfment receptors (STEP 2) and activate signaling pathways for the induction of phagocytosis. The culmination of this signal transduction is the generation of pseudopodia that help phagocytes surround and incorporate apoptotic cells (STEP 3). Materials engulfed exist as phagosomes, which subsequently fuse with the lysosomes for degradation (STEP 4) (Nainu, Shiratsuchi, and Nakanishi, 2017).Apoptotic Signaling Pathways
Apoptosis can be initiated through two main pathways: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway. Both pathways ultimately converge on the activation of caspases.
Intrinsic Pathway
The intrinsic pathway of apoptosis, also known as the mitochondrial pathway, is activated by a variety of internal stress signals, including severe DNA damage, oxidative stress, endoplasmic reticulum stress, and growth factor deprivation. These stressors lead to the activation of pro-apoptotic members of the Bcl-2 family, such as Bax and Bak, which oligomerize and insert into the mitochondrial outer membrane. This process, known as mitochondrial outer membrane permeabilization (MOMP), is a critical event leading to the release of key apoptogenic factors from the mitochondrial intermembrane space into the cytosol. Among these factors, cytochrome c plays a pivotal role by binding to apoptotic protease activating factor-1 (Apaf-1) in the presence of ATP, facilitating the assembly of a multi-protein complex known as the apoptosome. The apoptosome functions as a platform for the recruitment and activation of procaspase-9, which then undergoes autocatalytic cleavage to become active caspase-9. Caspase-9 then initiates a proteolytic cascade by cleaving and activating downstream executioner caspases such as caspase-3, caspase-6, and caspase-7. These executioner caspases are responsible for orchestrating the systematic degradation of cellular components, including cleavage of structural proteins, inactivation of DNA repair enzymes, and fragmentation of nuclear DNA, ultimately leading to the controlled disassembly and removal of the cell.
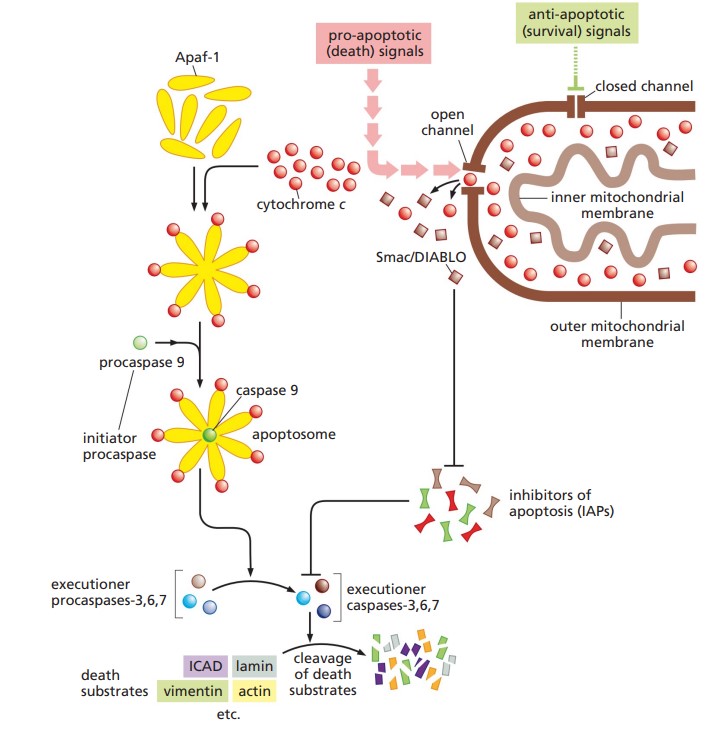 Fig. 6: Schema of apoptosis intrinsic pathway (The Biology of Cancer, Garland Science, 2nd Edition).
Fig. 6: Schema of apoptosis intrinsic pathway (The Biology of Cancer, Garland Science, 2nd Edition).Extrinsic Pathway
The extrinsic pathway of apoptosis is initiated through the interaction of specific extracellular death ligands with their cognate death receptors located on the cell surface. These death receptors, including Fas (CD95/Apo-1) and TNF receptor 1 (TNFR1), are integral members of the tumor necrosis factor receptor (TNFR) superfamily, characterized by the presence of a cytoplasmic death domain. Upon binding of death ligands, such as Fas ligand (FasL) or tumor necrosis factor-alpha (TNF-α), these receptors undergo trimerization, a conformational change that is essential for the subsequent recruitment of intracellular adaptor proteins. One critical adaptor protein is FADD (Fas-associated death domain), which binds to the death domains of the trimerized receptors via its own death domain.
FADD, in turn, recruits procaspase-8 through its death effector domain (DED), leading to the assembly of the death-inducing signaling complex (DISC). The close proximity of procaspase-8 molecules within the DISC facilitates their autocatalytic cleavage and activation. Activated caspase-8 acts as an initiator caspase, directly cleaving and activating downstream executioner caspases, such as caspase-3, caspase-6, and caspase-7. These executioner caspases are responsible for orchestrating the degradation of key structural and regulatory proteins, including nuclear lamins, cytoskeletal proteins, and DNA repair enzymes, culminating in the systematic disassembly of the cell.
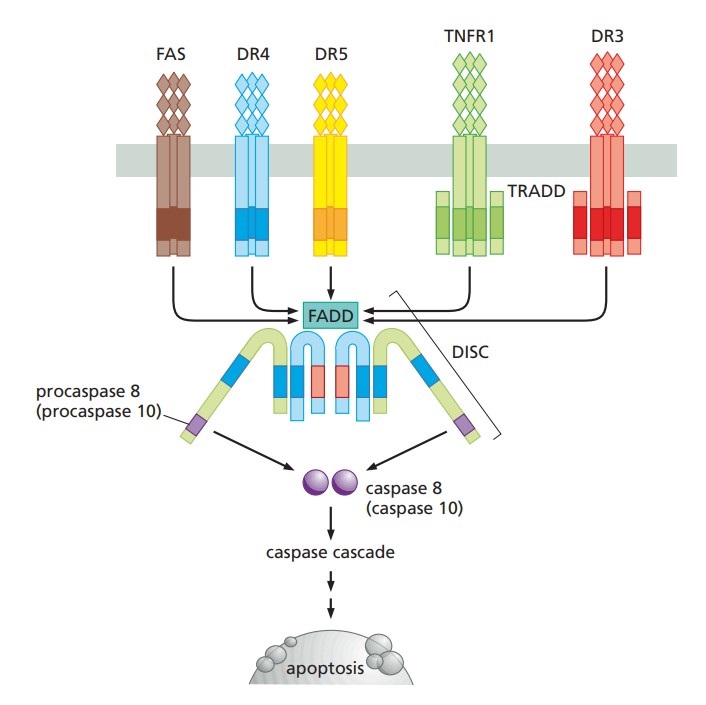 Fig. 7: Schema of apoptosis extrinsic pathway (The Biology of Cancer, Garland Science, 2nd Edition).
Fig. 7: Schema of apoptosis extrinsic pathway (The Biology of Cancer, Garland Science, 2nd Edition).Crosstalk Between Pathways
The intrinsic and extrinsic pathways are not completely independent and can influence each other. For example, activation of caspase-8 in the extrinsic pathway can cleave BID, leading to permeabilization of the mitochondrial outer membrane and activation of the intrinsic pathway.
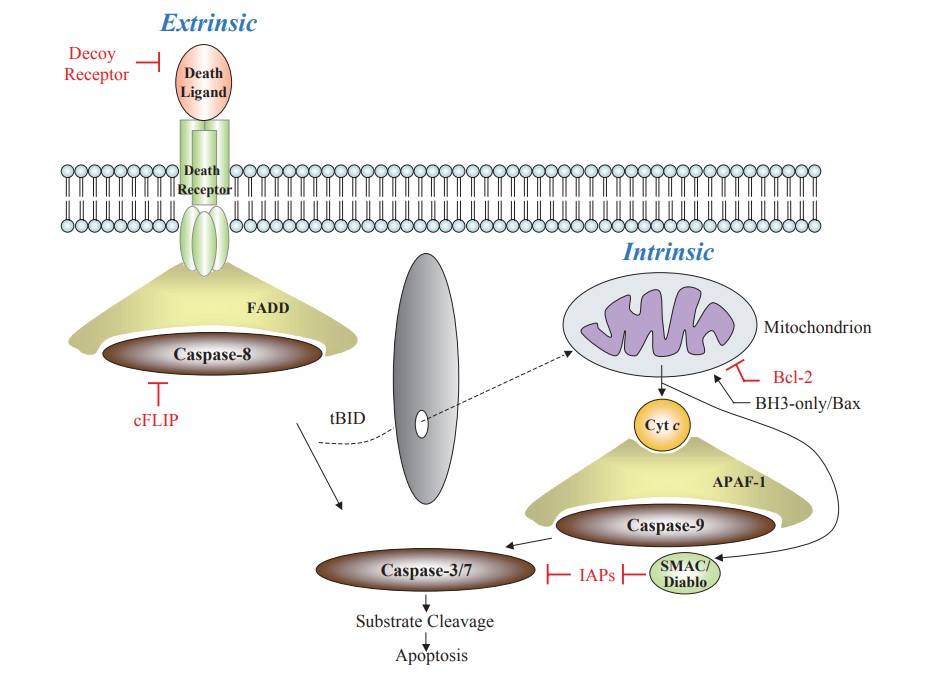 Fig. 8: Extrinsic and intrinsic cell death signaling pathways. The crosstalk between these two pathways exists via truncated BID (tBID) (Hattori et al., 2010).
Fig. 8: Extrinsic and intrinsic cell death signaling pathways. The crosstalk between these two pathways exists via truncated BID (tBID) (Hattori et al., 2010).Apoptosis and Diseases
Apoptosis plays a fundamental role in several physiological processes, including embryonic development, immune system regulation, and tissue remodeling. However, dysregulation of apoptosis can contribute to the pathogenesis of many diseases.
Cancer: One of the hallmarks of cancer is the evasion of apoptosis, allowing cancer cells to proliferate uncontrollably. Mutations in genes that regulate apoptosis, such as p53 or Bcl-2, are commonly observed in tumors. Therapies that restore the apoptotic response in cancer cells are a major area of research in oncology.
Neurodegenerative Diseases: Excessive apoptosis of neurons is implicated in neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases. In these conditions, abnormal protein aggregation and mitochondrial dysfunction can trigger apoptotic pathways, leading to neuronal loss.
Autoimmune Diseases: In autoimmune diseases, defective apoptosis can lead to the survival of autoreactive immune cells that attack the body's tissues. For example, in systemic lupus erythematosus (SLE), impaired clearance of apoptotic cells may contribute to the development of autoantibodies.
Infectious Diseases: Certain pathogens can manipulate apoptotic pathways to enhance their survival. For example, some viruses encode proteins that inhibit apoptosis, allowing infected cells to evade the immune response and continue to produce viral particles.
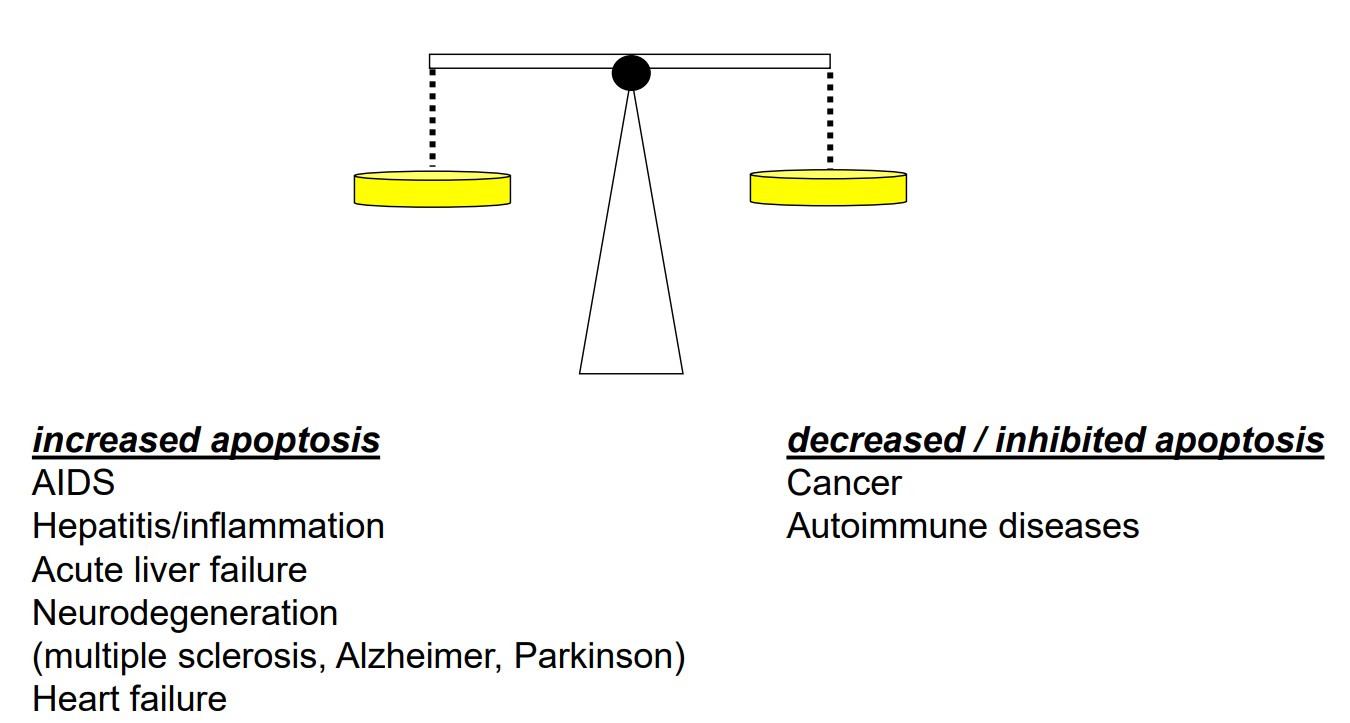 Fig. 9: Dysregulation of apoptosis can contribute to the pathogenesis of numerous diseases.
Fig. 9: Dysregulation of apoptosis can contribute to the pathogenesis of numerous diseases.Other Types of Programmed Cell Death
Historically, programmed cell death (PCD) has been considered equivalent to apoptosis. However, our current knowledge says that there are several types of programmed cell death depending on the cellular environment, external stress factors and yet unknown conditions, including apoptosis, necroptosis, ferroptosis, pyroptosis, parthanatos, autophagy-dependent cell death, each with different mechanisms and physiological implications. Here's a comparison between apoptosis and other types of PCD.
Necroptosis
Necroptosis is a form of programmed necrosis mediated by receptor-interacting protein kinases (RIPK1 and RIPK3) and the pseudokinase MLKL. It occurs when apoptosis is inhibited, often in response to death receptor signaling. Key morphologic features include cell swelling, organelle enlargement, plasma membrane rupture, and spillage of cellular contents, leading to inflammation. Activation of death receptors (e.g., TNFR1) in the absence of caspase-8 activity, certain viral infections, or chemical agents. Necroptosis leads to inflammatory cell death, often in response to infection or pathological conditions in which apoptosis is blocked.
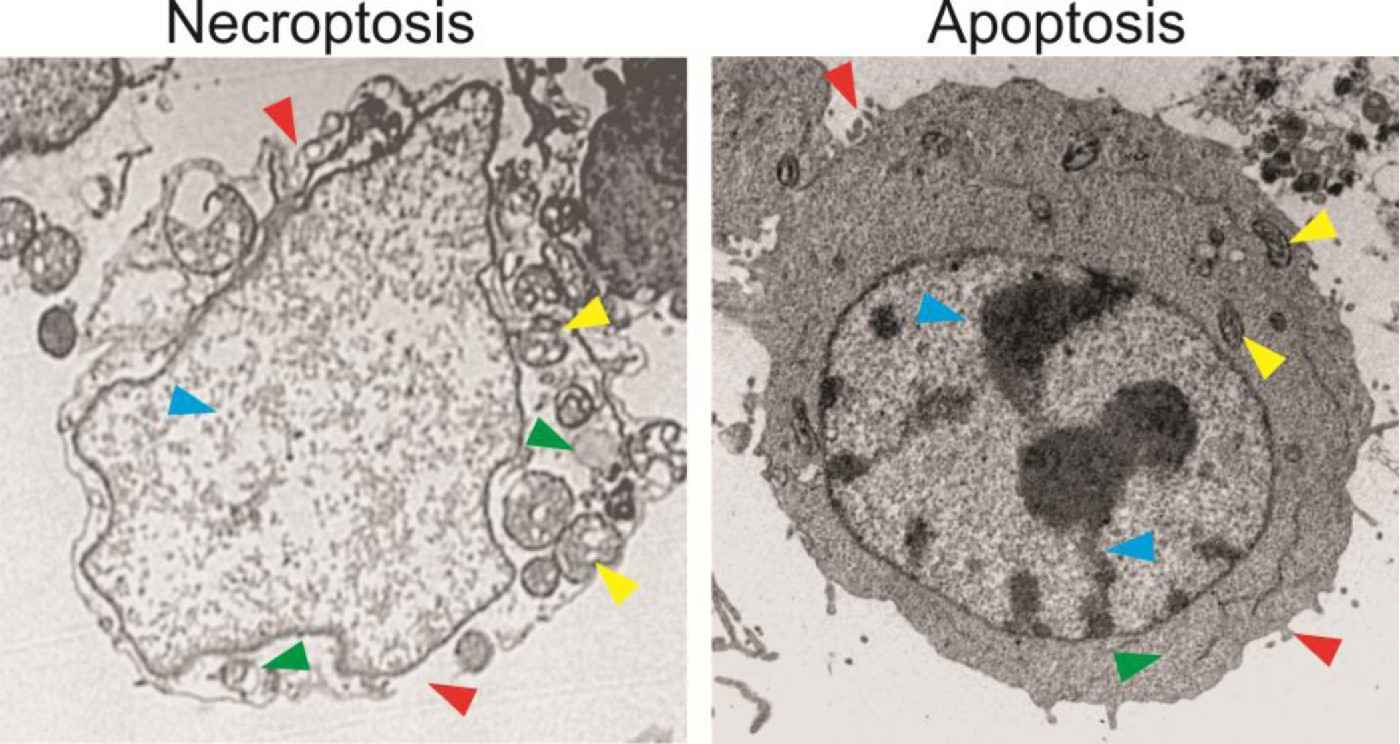 Fig. 10: HT29 colon cancer cells treated with an anticancer drug for 48 hours were analyzed by transmission electron microscopy. The cell undergoing necroptosis shows plasma membrane rupture and permeabilization, compared to the intact plasma membrane with blebbing in the apoptotic cell (red arrowheads). The necroptotic cell exhibits cytoplasm swelling and vacuolization, which are absent in the apoptotic cell (green arrowheads). The necroptotic cell has swelled mitochondria, in contrast to those in the apoptotic cells (yellow arrowheads). The necroptotic cell also lacks condensed and fragmented nuclei seen in the apoptotic cell (blue arrowheads) (Chen et al., 2016).
Fig. 10: HT29 colon cancer cells treated with an anticancer drug for 48 hours were analyzed by transmission electron microscopy. The cell undergoing necroptosis shows plasma membrane rupture and permeabilization, compared to the intact plasma membrane with blebbing in the apoptotic cell (red arrowheads). The necroptotic cell exhibits cytoplasm swelling and vacuolization, which are absent in the apoptotic cell (green arrowheads). The necroptotic cell has swelled mitochondria, in contrast to those in the apoptotic cells (yellow arrowheads). The necroptotic cell also lacks condensed and fragmented nuclei seen in the apoptotic cell (blue arrowheads) (Chen et al., 2016).Ferroptosis
Ferroptosis is an iron-dependent form of cell death characterized by the accumulation of lipid peroxides leading to lethal membrane damage. Unlike other forms of cell death, ferroptosis is characterized by the absence of typical apoptotic or necrotic features, with small mitochondria, condensed mitochondrial membranes, and ruptured plasma membranes. Ferroptosis is induced by iron overload, inhibition of the cystine/glutamate antiporter (system xc-), depletion of glutathione, or inhibition of glutathione peroxidase 4 (GPX4). Ferroptosis has been implicated in several diseases, including neurodegeneration, cancer, and ischemia-reperfusion injury.
Pyroptosis
Pyroptosis is a caspase-1 dependent cell death process often associated with inflammation. It is initiated by the activation of inflammasomes, leading to caspase-1 activation, which cleaves gasdermin D, forming pores in the membrane. Similar to necroptosis, pyroptosis involves rapid cell swelling, membrane rupture, and release of pro-inflammatory cytokines (e.g., IL-1β, IL-18). Triggers are recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs). Pyroptosis results in an inflammatory response that helps fight infection but, if dysregulated, can contribute to sepsis or chronic inflammatory diseases.
Parthanatos
Parthanatos is driven by the overactivation of poly(ADP-ribose) polymerase-1 (PARP-1), leading to the translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus, causing large-scale DNA fragmentation. Parthanatos causes chromatin condensation and DNA fragmentation without caspase activation. Unlike apoptosis, parthanatos does not involve the formation of apoptotic bodies. It is triggered by extensive DNA damage, oxidative stress or excitotoxicity (especially in neurons). Parthanatos has been implicated in neurodegenerative diseases, stroke, and other conditions associated with excessive DNA damage and PARP-1 activation.
Autophagy-Dependent Cell Death
Autophagy-dependent cell death occurs when autophagy, a process typically associated with cell survival through the degradation of cellular components, becomes excessive or dysregulated, leading to cell death. Autophagy-dependent cell death is characterized by extensive autophagosome formation, degradation of cellular organelles, and lysosomal membrane permeabilization. It does not involve caspase activation. Triggers include nutrient deprivation, hypoxia, or treatment with certain drugs that induce excessive autophagy. Autophagy-dependent cell death can occur in several diseases, including cancer, neurodegeneration, and heart disease, where autophagy contributes to cell death rather than survival.
Case Study
Case 1: Soengas, M. S.; et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature, 2001; 409(6817), 207–211.
Apaf-1 is a cell death effector that cooperates with cytochrome c and caspase-9 to mediate p53-dependent apoptosis. In this study, the researchers showed that metastatic melanomas often lose Apaf-1, the spread of primary melanoma cells to distant organs such as nonregional lymph nodes, lung, liver, brain and bone. Apaf-1 is inactivated in metastatic melanomas, leading to defects in the execution of apoptotic cell death. Loss of Apaf-1 expression is associated with allelic loss in metastatic melanoma.
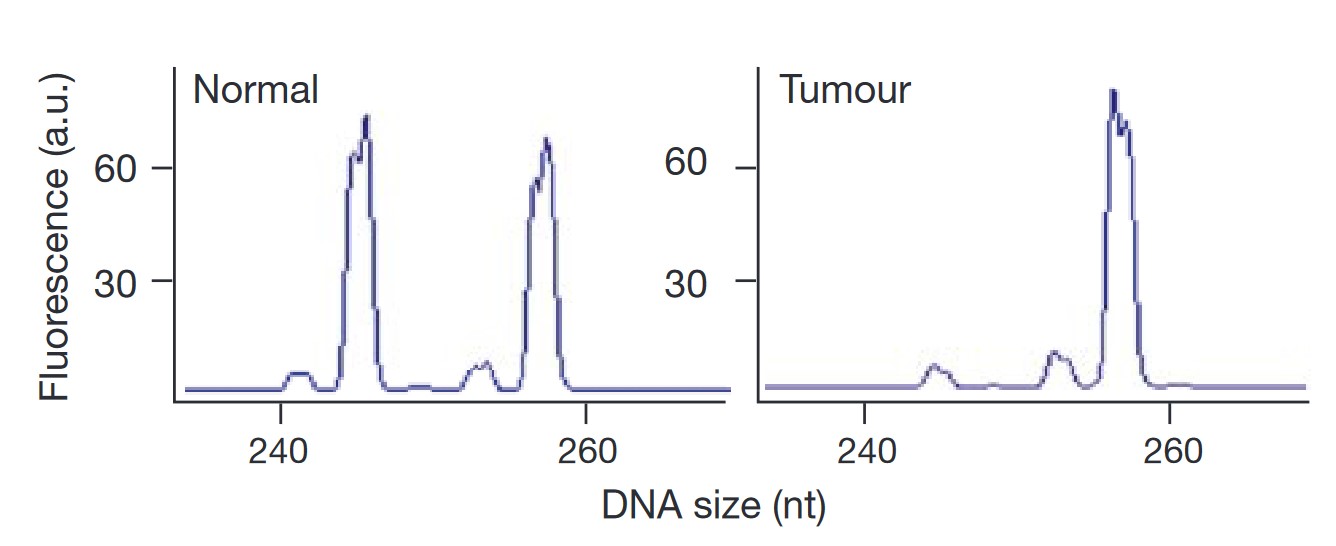 Fig. 11: Apaf-1 loss in metastatic melanoma. Fluorescent electropherograms for microsatellite marker D12S393 on chromosome 12q22–23. The normal (left) and tumor (right) counterparts of case four are indicated. a.u., arbitrary units.
Fig. 11: Apaf-1 loss in metastatic melanoma. Fluorescent electropherograms for microsatellite marker D12S393 on chromosome 12q22–23. The normal (left) and tumor (right) counterparts of case four are indicated. a.u., arbitrary units.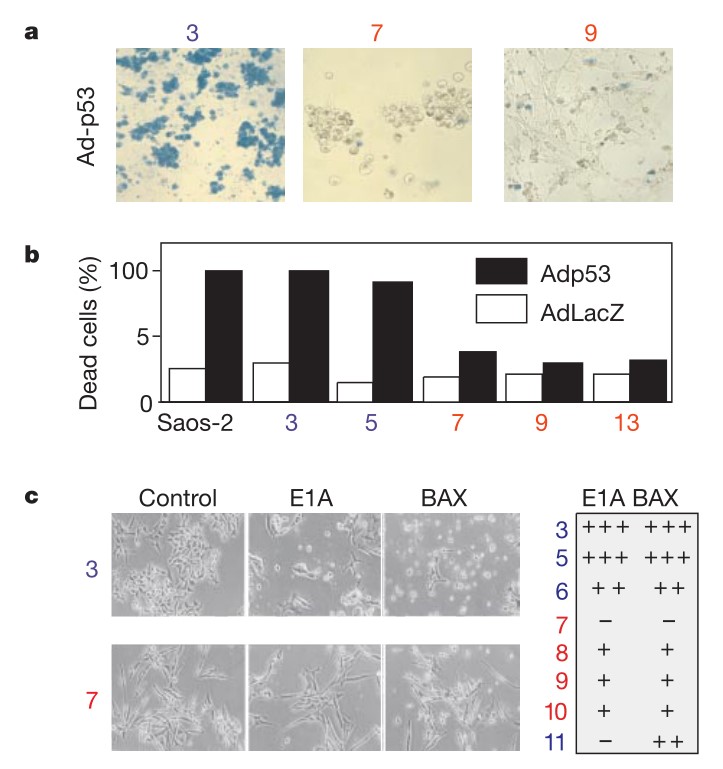 Fig. 12: Apoptotic defects in Apaf-1-negative melanoma cells. a, Viability measurements of melanoma cells after infection with a replication-defective adenovirus expressing p53 (Adp53) or a control LacZ. Seventy-two hours after infection, representative cultures were stained with trypan blue to visualize dead cells. b, Quantification of data in a. Saos-2 cells are included as a positive control28. c, Viability of representative melanoma lines 36 h after introduction of E1A or BAX by retrovirus-mediated gene transfer. Photomicrographs of two representative cell lines are indicated, with quantification of data from a larger series shown to the right. Apaf-1-positive cell lines are indicated in blue, and Apaf-1negative cell lines are indicated in red.
Fig. 12: Apoptotic defects in Apaf-1-negative melanoma cells. a, Viability measurements of melanoma cells after infection with a replication-defective adenovirus expressing p53 (Adp53) or a control LacZ. Seventy-two hours after infection, representative cultures were stained with trypan blue to visualize dead cells. b, Quantification of data in a. Saos-2 cells are included as a positive control28. c, Viability of representative melanoma lines 36 h after introduction of E1A or BAX by retrovirus-mediated gene transfer. Photomicrographs of two representative cell lines are indicated, with quantification of data from a larger series shown to the right. Apaf-1-positive cell lines are indicated in blue, and Apaf-1negative cell lines are indicated in red.References
- Chen, D., Yu, J., & Zhang, L. (2016). Necroptosis: An alternative cell death program defending against cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 1865(2), 228–236.
- Elmore, S. (2007). Apoptosis: A review of programmed cell death. Toxicologic Pathology, 35(4), 495–516.
- Fuchs, Y., & Steller, H. (2011). Programmed cell death in animal development and disease. Cell, 147(4), 742–758.
- Green, D. R., & Llambi, F. (2015). Cell death signaling. Cold Spring Harbor Perspectives in Biology, 7(12), a006080.
- Hattori, Y., Takano, K., Teramae, H., Yamamoto, S., Yokoo, H., & Matsuda, N. (2010). Insights into sepsis therapeutic design based on the apoptotic death pathway. Journal of Pharmacological Sciences, 114(4), 354–365.
- Mattson, M. P. (2000). Apoptosis in neurodegenerative disorders. Nature Reviews. Molecular Cell Biology, 1(2), 120–129.
- Nainu, F., Shiratsuchi, A., & Nakanishi, Y. (2017). Induction of apoptosis and subsequent phagocytosis of virus-infected cells as an antiviral mechanism. Frontiers in Immunology, 8.
- Reed, J. C. (2000). Mechanisms of apoptosis. The American Journal of Pathology, 157(5), 1415–1430.
- Soengas, M. S., Capodieci, P., Polsky, D., Mora, J., Esteller, M., Opitz-Araya, X., McCombie, R., Herman, J. G., Gerald, W. L., Lazebnik, Y. A., Cordón-Cardó, C., & Lowe, S. W. (2001). Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature, 409(6817).
- Strasser, A., Jost, P. J., & Nagata, S. (2009). The many roles of FAS receptor signaling in the immune system. Immunity, 30(2), 180–192.