Protein Phylogenetic Analysis
Background
Introduction
Unlocking Evolutionary Insights: Comprehensive Protein Phylogenetic Analysis Services
Protein phylogenetic analysis is pivotal for decoding evolutionary relationships, tracing gene family origins, and identifying functional adaptations in modern biological research. By integrating evolutionary genetics with multiple sequence alignment, this approach reveals conserved domains and mutation patterns critical for drug discovery, species classification, and understanding disease mechanisms.
At Creative BioMart , we specialize in delivering end-to-end bioinformatics services powered by cutting-edge algorithms. Our team offers precise phylogenetic tree construction, leveraging decades of expertise to ensure accuracy in homology modeling, divergence time estimation, and ancestral sequence reconstruction. Customizable workflows cater to diverse research goals, from academic studies to industrial R&D.
Our unique value lies in merging advanced protein phylogenetic analysis tools with custom bioinformatics consultation. Clients gain actionable insights through tailored data interpretation, interactive visualizations, and strategic guidance to optimize experimental design. Partner with Creative BioMart to accelerate your evolutionary studies with scalable, publication-ready solutions.
Overview of Protein Phylogenetic Analysis
Phylogenetic Analysis in Protein Evolution: Decoding Molecular Relationships
Protein phylogenetic analysis examines evolutionary relationships among proteins, tracing ancestry and functional divergence through molecular phylogenetics. This method identifies conserved domains and adaptive mutations, aiding in drug design, species classification, and disease mechanism studies.
By aligning sequences and constructing a phylogenetic tree, researchers infer evolutionary timelines and functional similarities. For instance, homologous proteins with shared nodes suggest common ancestry, while distant branches highlight functional specialization—critical for predicting protein interactions or engineering enzymes.
Our infographic visually maps a phylogenetic tree, simplifying complex evolutionary data. Clustering patterns and bootstrap values clarify confidence in branching events, enabling researchers to pinpoint key evolutionary shifts. Integrate these insights with genomic data to advance studies in protein evolution and adaptive biology.
Service Details
Creative BioMart offers efficient, customizable bioinformatics services (e.g., genome evolution modeling, repeat detection) using high-throughput analysis pipelines. With modular workflows and tailored parameters (alignment thresholds, substitution models), projects typically conclude within 24-48 hours, balancing speed and precision for computational biology needs.

Core Services
|
Methodologies |
Applications |
|---|---|
|
Multiple Sequence Alignment (MSA) |
Using tools like Clustal Omega and MAFFT, we align protein/DNA sequences to identify conserved regions, mutations, and structural motifs. This step is critical for accurate phylogeny inference and functional annotation in evolutionary studies. |
|
Phylogeny Inference Programs |
Our pipelines apply maximum likelihood, Bayesian frameworks, and the DNA parsimony algorithm to reconstruct evolutionary trees. These methods reduce homoplasy errors, resolve polytomies, and validate branching patterns via bootstrapping for reliable evolutionary insights. |
|
DNA Parsimony Algorithm |
This algorithm minimizes evolutionary steps to infer ancestral relationships, ideal for resolving closely related species. It complements probabilistic methods, ensuring robust tree topology validation. |
|
Evolutionary Genetics Analysis |
We calculate selection pressures (e.g., dN/dS ratios), detect adaptive mutations, and model population dynamics. Combined with genome evolution simulation, this reveals mechanisms like positive selection or genetic drift. |
|
Genome Evolution Simulation |
Simulate scenarios (e.g., gene duplication, horizontal transfer) to test evolutionary hypotheses. Customizable parameters allow modeling adaptive radiation or extinction events, aiding experimental design. |
|
Protein Family Classification |
Leveraging domain architecture analysis and machine learning, we classify orthologs/paralogs into families. This supports functional prediction, pathway analysis, and comparative genomics. |
|
De Novo Repeat Detection |
Using k-mer-based tools and HMMs, we identify novel repetitive elements (e.g., transposons) in genomes, crucial for studying genome stability, regulatory networks, and evolutionary plasticity. |
Service Delivery & Submission Guidelines
-
- What You Receive:
Fast, Transparent Results: Receive detailed reports (PDF/Excel), interactive phylogenetic trees (Newick/NEXUS), and alignment files within 1-7 days, tailored to your project’s complexity.
Long-Term Access: All raw data, analysis scripts, and quality control metrics are securely stored for 6 months for your convenience. -
- What We Need from You:
Sequence Data: FASTA, VCF, or BLAST files (clearly labeled).
Project Context: Species/strain details, sequencing methods, and research objectives (e.g., evolutionary divergence study).
Custom Preferences: Specify parameters like substitution models or bootstrap replicates—or let our experts optimize them for you.
Why Choose Us?
- Proven Expertise & Experience
With 15+ years in bioinformatics and 1,000+ projects delivered, our PhD-led team excels in complex tasks like phylogenetic analysis and genome annotation, ensuring precision for academic and industrial clients. - Uncompromising Quality Assurance
Multi-tier validation (peer review, algorithmic checks) guarantees data integrity. Our workflows adhere to global standards (ISO-certified processes), minimizing errors in high-throughput analysis or evolutionary modeling. - Tailored Solutions
Adjust parameters (e.g., substitution models, alignment thresholds) and output formats to align with your goals—whether prioritizing speed for genome assembly or depth for protein family classification. - End-to-End Support
Dedicated 24/7 assistance from project design to final delivery. We troubleshoot technical challenges and optimize workflows, ensuring deadlines are met without sacrificing accuracy.
Case Study
Case 1: Phylogenetic Analysis of the Vascular Endothelial Growth Factor (VEGF) Family
Research Background : The VEGF family plays a crucial role in angiogenesis and vascular development. Understanding the evolutionary relationships within this family can provide insights into their functional diversity.
Methods :
- Sequence Alignment: Multiple sequence alignment using Clustal Omega.
- Phylogenetic Tree Construction: Maximum likelihood method was employed to construct the phylogenetic tree.
Results : The study revealed high conservation among VEGF family members across vertebrates, with some lineage-specific variations that may indicate functional divergence.
Significance : This research enhances the understanding of the evolutionary history of the VEGF family, which is essential for developing therapeutic strategies targeting angiogenesis.
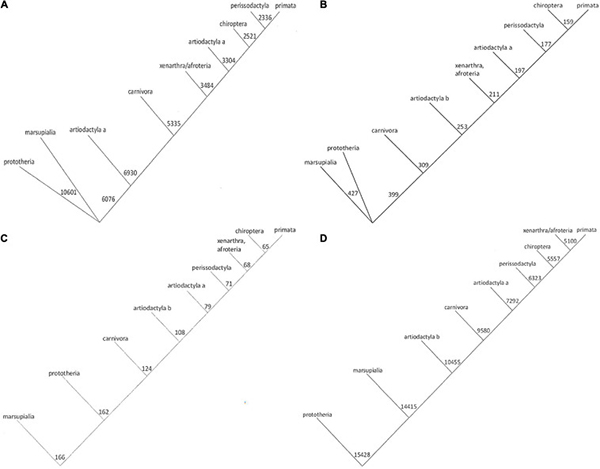
Fig1. Phylogenetic trees for Osteopontin evolution. (Wang, et al ., 2021)
Case 2: Phylogenetic Analysis of Amino Acid Replacement in Proteins
Research Background : Amino acid substitutions are fundamental to protein evolution. Studying these substitutions helps elucidate the evolutionary constraints and dynamics of protein families.
- Sequence Collection: Amino acid sequences from various protein families were collected.
- Phylogenetic Analysis: Empirical profile mixture models were used for phylogenetic reconstruction.
Results : The study identified specific patterns of amino acid replacements that are influenced by both structural and functional constraints of proteins.
Significance : This research provides a deeper understanding of the evolutionary processes shaping protein sequences and structures.
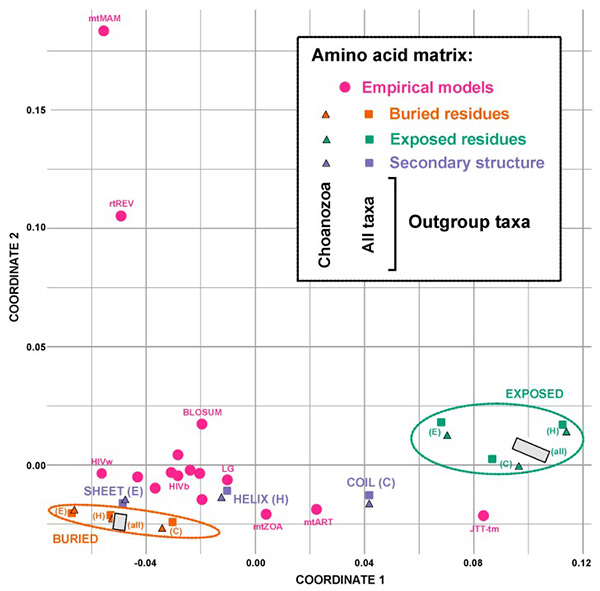
Fig2. Multidimensional scaling plot showing the Euclidean distances between various amino acids exchange rate matrices. (Pandey, et al ., 2020)
Case 3: Evolutionary Origins of BACE1 Proteolytic Function in Alzheimer's Disease
Research Background : Alzheimer's disease is characterized by the accumulation of β-amyloid (Aβ) in the brain, produced by the proteolysis of amyloid precursor protein (APP) by β-secretase (BACE1). BACE1 is a key therapeutic target, and understanding its evolutionary history is crucial for developing treatments.
Methods :
- Sequence Collection: BACE1 orthologs were examined across early-diverging animal lineages, including cnidarians, ctenophores, and choanoflagellates.
- Phylogenetic Analysis: The evolutionary relationships of BACE1 and BACE2 genes were analyzed.
- Functional Assays: The ability of BACE1 orthologs to cleave APP and release Aβ was tested.
Results : The study found that BACE1 likely evolved from a gene duplication event near the base of the animal clade. The specific proteolytic function of BACE1, capable of cleaving APP to release Aβ, emerged during early animal diversification, hundreds of millions of years before the evolution of the APP/Aβ substrate. The most basal BACE1 ortholog was identified in cnidarians (Hydra), which retained the ability to cleave APP and release Aβ. In contrast, BACE1/2 genes from more divergent lineages, such as ctenophores (Mnemiopsis) and choanoflagellates (Monosiga), lacked this specific proteolytic activity.
Significance: This research provides insights into the evolutionary origins of BACE1's proteolytic function, suggesting it emerged early in animal evolution, likely following a gene duplication event. Understanding this evolutionary history can inform the development of therapeutic strategies targeting BACE1 in Alzheimer's disease.
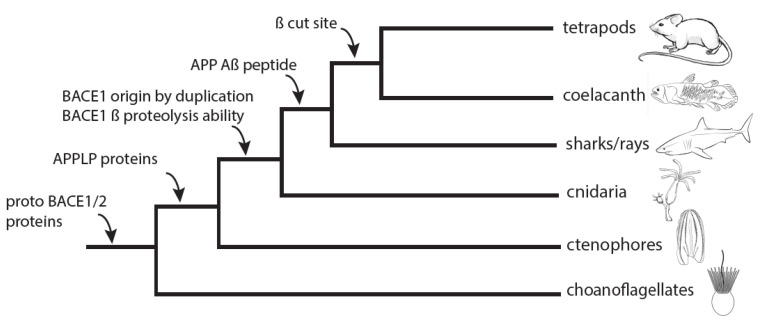
Fig3. Relative timeline for BACE1 and Aβ evolution. (Langeland, et al ., 2024)
Customer Testimonials
-
"Creative BioMart’s phylogenetic analysis expertise accelerated our antibody evolution project. Their custom workflows and 24/7 support ensured seamless integration with our R&D timelines."
-Dr. Sarah Johnson, Head of Biotherapeutics | Biopharma Innovator
-
"Partnering with them for genome annotation saved months of manual work. Their ISO-certified quality control and adaptive pipelines delivered publication-ready data."
-Prof. Michael Tan, Evolutionary Genomics Lab
-
"From high-throughput analysis to repeat detection, their team optimized parameters for crop genome studies. Fast turnaround without compromising accuracy."
-Dr. Lisa Park, Senior Bioinformatician | AgriGenome Solutions
-
"Custom protein family classification enabled precise drug target identification. Their computational biology tools and post-analysis support were invaluable."
-Dr. Raj Patel, CSO | NeuroBio Therapeutics
-
"Trust their evolutionary genetics analysis for non-model species. Rigorous validation and tailored reporting streamlined our marine biodiversity research."
-Dr. Emily Wu, Director | Marine Genomics Institute
-
"Outsourced de novo genome assembly with confidence. Their expertise in handling complex datasets and transparent communication exceeded expectations."
-Dr. Carlos Mendez, Lead Researcher
FAQs
-
Q: What types of protein data can you analyze?
A: We analyze FASTA, BLAST, or VCF files for single proteins or entire families. Our pipelines support multiple sequence alignment and phylogenetic tree construction for evolutionary, structural, or functional studies.
-
Q: How do you ensure phylogenetic tree accuracy?
A: We use validated algorithms (RAxML, MrBayes) and rigorous QC steps, including bootstrap/posterior probability checks and manual peer review, to minimize errors in phylogeny inference.
-
Q: Can you customize analyses for specific research goals?
A: Absolutely. Specify parameters (substitution models, divergence dating) or let our experts design workflows for positive selection detection or protein family classification.
-
Q: How secure is our submitted data?
A: All data is encrypted during transfer/storage, with strict confidentiality agreements. Clients retain full ownership, and files are purged post-project unless requested otherwise.
-
Q: Can your analysis integrate with other bioinformatics services?
A: Yes. Combine protein phylogenetic analysis with downstream services like functional annotation or 3D structure prediction for comprehensive insights into protein evolution and interactions .
Other Resources
References:
- Wang X, Weber GF. Quantitative Analysis of Protein Evolution: The Phylogeny of Osteopontin. Front Genet. 2021;12:700789.
- Pandey A, Braun EL. Phylogenetic Analyses of Sites in Different Protein Structural Environments Result in Distinct Placements of the Metazoan Root. Biology (Basel). 2020;9(4):64.
- Langeland JA.; et al. Early Animal Origin of BACE1 APP/Aβ Proteolytic Function. Biology (Basel). 2024;13(5):320.
Contact us or send an email at for project quotations and more detailed information.
Quick Links
-

Papers’ PMID to Obtain Coupon
Submit Now -

Refer Friends & New Lab Start-up Promotions