Uncategorized Monday, 2024/07/01
In a new study, researchers from research institutions such as Baylor School of Medicine revealed the molecular events that lead to osteogenesis imperfecta type V. This discovery provides new possibilities for designing treatments for this incurable disease. The relevant research results were published online in the Journal of Clinical Investigation, under the title "The IFITM5 mutation in osteogenesis imperfecta type V is associated with an ERK/SOX9-dependent osteoprogenitor differentiation defect".
V-type osteogenesis imperfecta is a brittle bone disease caused by a mutation in the IFITM5 gene. This genetic mutation hinders the normal development of backbone cells into mature cells that form healthy bones. This mutation can lead to the formation of extremely brittle bones. Children with this disease may experience recurrent fractures, skeletal deformities, chronic pain, and other complications.
Our Featured Products
| Cat No. | Product Name | Source | Species | Tag |
| FITM5-1251H | Recombinant Human IFITM5 | Mammalian Cell | Human | His |
| IFITM5-1193H | Recombinant Human IFITM5 Protein, MYC/DDK-tagged | HEK293 | Human | Myc/DDK |
| IFITM5-1146H | Recombinant Human IFITM5 Protein, His (Fc)-Avi-tagged | HEK293 | Human | His (Fc)-Avi |
| IFITM5-1853HFL | Recombinant Full Length Human IFITM5 Protein, C-Flag-tagged | Mammalian cells | Human | Flag |
| SOX9-2415H | Recombinant Human SOX9, GST-tagged | E.coli | Human | GST |
| SOX9-30694TH | Recombinant Human SOX9 | E.coli | Human | N/A |
| SOX9-175HFL | Active Recombinant Full Length Human SOX9 Protein, C-Flag-tagged | Mammalian cells | Human | Flag |
| SOX9-3043H | Recombinant Human SOX9 Protein, MYC/DDK-tagged | HEK293 | Human | Myc/DDK |
| SOX9-2078H | Recombinant Human SOX9 Protein, His (Fc)-Avi-tagged | HEK293 | Human | His (Fc)-Avi |
Dr. Brendan Lee, Professor of Molecular and Human Genetics at Baylor School of Medicine, said, "Crispy bone disease, also known as osteogenesis imperfecta, is a rare group of diseases that affect connective tissue - tissues such as bones support and protect other tissues in the human body. Most types of osteogenesis imperfecta are caused by genetic mutations that disrupt collagen synthesis or processing, but not V-type osteogenesis imperfecta."
Dr. Ronit Marom, Assistant Professor of Molecular and Human Genetics at Baylor School of Medicine, said, "V-type osteogenesis imperfecta is unique because all patients have identical mutations in the IFITM5 gene. This mutation leads to the production of a longer IFITM5 protein; however, the biological function of this protein in the bone and the reasons for this mutation causing osteogenesis imperfecta are not very clear."
These authors genetically modified mice to express the mutated IFITM5 gene at certain stages of skeletal development, thereby establishing an animal model of V-type osteogenesis imperfecta. These genetically modified mice reproduced most of the characteristics of human V-type osteogenesis imperfecta, enabling analysis of its underlying molecular mechanisms.
They found that mutations in the IFITM5 gene play a role at the level of bone marrow cells, altering the normal processes that lead to bone formation. Lee said, "Bone marrow cells play a dominant role in bone development and the formation of bone healing after fractures - first, they produce cartilage, and then turn into bones."
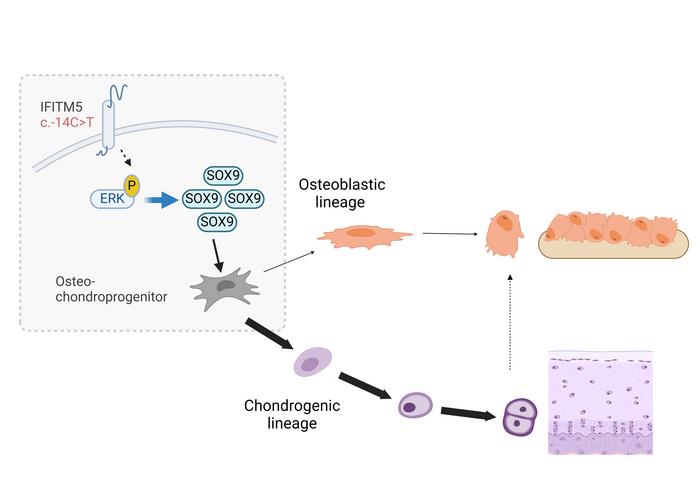 The IFITM5 mutation in mice disrupted this process. Osteopogenic cells did not transform from cartilage to bone, but instead formed overgrown cartilage calluses in the areas where new bones should have formed.
The IFITM5 mutation in mice disrupted this process. Osteopogenic cells did not transform from cartilage to bone, but instead formed overgrown cartilage calluses in the areas where new bones should have formed.
Lee said, "Our findings help explain what we see in patients with V-type osteogenesis imperfecta. Their bones are not only prone to breakage, but when the stem cells try to heal them, they form large cartilage callus instead of bone. This is like the stem cells not completing their work and getting stuck in a cycle, preferring to form cartilage rather than mature into bone. So far, we believe that osteogenesis imperfecta is the result of abnormal bone development. Excitingly, we have found that V-type osteogenesis imperfecta is actually a common result of abnormal differentiation of stem cells, leading to an imbalance in cartilage and bone development."
These authors also identified two main molecular roles that drive this bone maturation defect. Marom said, "The ERK/MAP kinase signaling pathway and transcription factor SOX9 are significantly increased. Interestingly, when we inactivate ERK/MAP kinase signaling or SOX9 through drug or genetic methods, we are able to restore normal bone development in mutation models. These findings not only provide information on the pathogenesis of V-type osteogenesis imperfecta, but also promote the development of future therapies for this disease."
Lee said, "In addition, all patients with V-type osteogenesis imperfecta have the same IFITM5 mutation, which may be an advantage in developing gene therapies targeting this mutated gene. A gene therapy for treating IFITM5 gene mutations will be applicable to all patients."
This study is another example of the value of rare disease research, which helps to improve understanding and treatment of common diseases. Lee said, "Understanding how V-type osteogenesis imperfecta occurs provides new insights into similar but more common skeletal diseases such as osteoporosis, and may also improve existing treatment methods."
Reference Ronit Marom et al. The IFITM5 mutation in osteogenesis imperfecta type V is associated with an ERK/SOX9-dependent osteoprogenitor differentiation defect. Journal of Clinical Investigation, 2024, doi:10.1172/JCI170369.
Related Products and Services
CD Antigens Targets of CAR-T Cell Therapy Cytokines Cancer Drug Targets Immune Checkpoint Proteins Protein Interaction Service Protein Expression and Purification Services Drug Discovery Screening Protein Pathway Profiling