Uncategorized Tuesday, 2024/03/19
Researchers from the University of Toronto published a research paper titled “Ex vivo activation of the GCN2 pathway metabolically reprograms T cells, leading to enhanced adoptive cell therapy” in the Cell Reports Medicine journal.
This study confirms that in vitro activation of the GCN2 pathway can metabolize reprogrammed T cells, thereby enhancing the therapeutic effect of adoptive cell therapy.
Featured Products
Manipulating T cell metabolism has become an attractive way to enhance the efficacy of adoptive cell therapy (ACT). Previous studies have shown that in preclinical models, enhancing mitochondrial oxidative phosphorylation (OXPHOS) of T cells through drugs or genes can improve tumor control. Interestingly, recent studies have found that transient glucose starvation in vitro can also lead to metabolic reprogramming of T cells and better tumor control. These data suggest that evolutionarily conserved metabolic stress pathways may be an attractive target for improving the efficacy of ACT.
GCN2 kinase is an important sensor for amino acid starvation. Once activated, GCN2 will release a large amount of proteins, including TNF-α, IL-6, and IL-10. These proteins can promote cell growth and division, thereby enhancing the immune response ability of cells. GCN2 phosphorylates eukaryotic initiation factor 2a and induces protein translation reprogramming, thereby generally inhibiting global protein translation, while promoting the expression of transcription activating factor 4 (ATF4) and other transcription factors involved in cell autophagy and protein uptake induction to coordinate the program of gene and protein expression, known as comprehensive stress response (ISR).
The role of GCN2-ATF4-induced comprehensive stress response (ISR) in CD8+T cells and anti-tumor immunity is still unclear. Early studies have shown that indoleamine 2,3-dioxygenase 1 (IDO1) activated leading to tryptophan depletion and GCN2 activation, inhibiting the function and proliferation of CD8+T cells. On the contrary, T cells lacking GCN2 exhibit proliferation defects during activation. In a mouse glioma model, CD8+T cells lacking GCN2 exhibit impaired anti-tumor immunity. However, in the B16 melanoma model, the specific absence of GCN2 did not affect anti-tumor immunity.
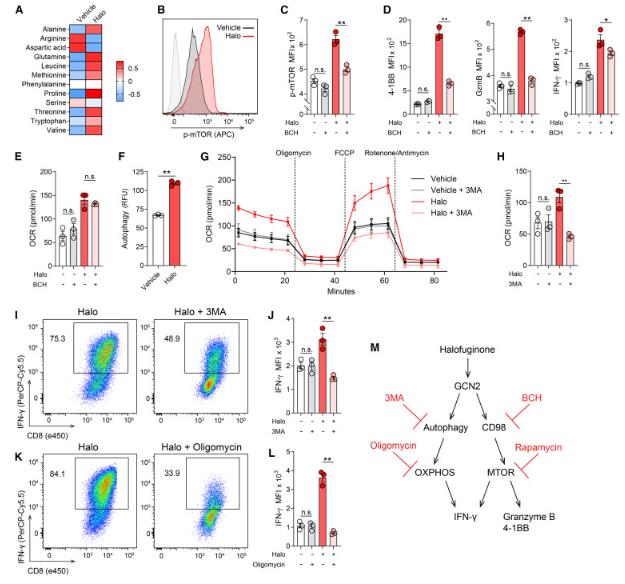
Given the conflicting research findings mentioned above, the research team attempted to determine the impact of activation of the GCN2 pathway on the metabolism, effector function, and anti-tumor ability of effector CD8+T cells.
In this latest study, the research team found that using the GCN2 agonist halofuginone (halo) to activate the amino acid starvation response of effector CD8+T cells in vitro can enhance their oxidative metabolism and effector function.
Mechanistically, this study identified autophagy and the CD98-mTOR signaling axis as key downstream mediators of the phenotype induced by halo therapy. The adoption and transfer of halo-treated CD8+T cells into a tumor-bearing mouse model resulted in a strong tumor control and healing response. In vivo, halo-treated T cells synergistically interact with 4-1BB agonist antibodies, inhibiting tumor growth in a mouse model resistant to immunotherapy. Importantly, the use of halo to treat human CD8+T cells has also led to similar metabolic and functional reprogramming.
These findings indicate that activating amino acid starvation response with GCN2 agonist halofuginone (halo) can enhance T cell metabolism and anti-tumor activity.
Related Products and Services
Cytokines Cancer Drug Targets Immune Checkpoint Proteins Protein Interaction Service Protein Expression and Purification Services Drug Discovery Screening Protein Pathway Profiling
Reference St Paul M, Saibil SD, Kates M, Han S, Lien SC, Laister RC, Hezaveh K, Kloetgen A, Penny S, Guo T, Garcia-Batres C, Smith LK, Chung DC, Elford AR, Sayad A, Pinto D, Mak TW, Hirano N, McGaha T, Ohashi PS. Ex vivo activation of the GCN2 pathway metabolically reprograms T cells, leading to enhanced adoptive cell therapy. Cell Rep Med. 2024 Mar 1:101465. doi: 10.1016/j.xcrm.2024.101465. Epub ahead of print. PMID: 38460518.